


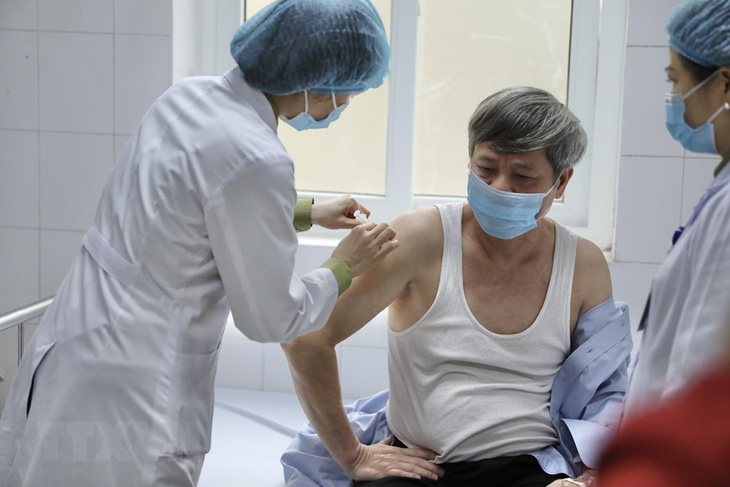
Thứ trưởng Bộ Khoa học và Công nghệ Phạm Công Tạc tiêm thử nghiệm vắc xin Nano Covax giai đoạn 2 tại Học viện Quân y (Bộ Quốc Phòng) sáng 26-3 - Ảnh: TTXVN
Cùng với đó, có rất nhiều thắc mắc từ dư luận đã đặt ra: Nanogen kiến nghị cấp phép khẩn cấp cho vắc xin Nano Covax có "nóng vội" hay không? Quy chuẩn khoa học để cấp phép một loại vắc xin trong điều kiện khẩn cấp hiện nay như thế nào?
Tuổi Trẻ đã trao đổi với đại diện nhà sản xuất, Bộ Y tế (cơ quan quản lý chuyên môn, cấp phép) và ý kiến của các chuyên gia về vấn đề này để có thêm thông tin.
Gặp khó khăn với cơ chế "xin - cho"?
Sáng 23-6, Công ty cổ phần công nghệ sinh học dược Nanogen (gọi tắt là Nanogen) đã có những chia sẻ với báo chí sau khi kiến nghị cấp phép khẩn cấp cho vắc xin Nano Covax mà đại diện Bộ Y tế đánh giá là "nóng vội, chưa đủ dữ liệu khoa học".
Theo đại diện Nanogen, đến nay quá trình thử nghiệm đang bước vào giai đoạn 3 (giai đoạn cuối cùng) với quy mô thử nghiệm trên 13.000 người (đã có 1.000 người tiêm thử nghiệm mũi 1). Việc gửi kiến nghị cấp phép khẩn cấp cho vắc xin lên Thủ tướng, Nanogen lý giải là "thể hiện sự mong muốn lớn nhất của đơn vị", đồng thời "thay mặt cho người dân để sớm có vắc xin phòng COVID-19, an tâm quay trở lại cuộc sống".
Đại diện Nanogen cho rằng đơn vị gặp nhiều khó khăn với cơ chế "xin - cho" trong quá trình nghiên cứu, thử nghiệm, cấp phép qua các giai đoạn của vắc xin Nano Covax. Điển hình như hồi tháng 4-2021 khi hoàn tất thử nghiệm giai đoạn 2, Nanogen xin cho thực hiện giai đoạn 3 nhưng Bộ Y tế không đồng ý và phải chờ đợi đến ngày 10-6 mới bắt đầu.
"Đầu tư sản xuất, thử nghiệm một loại vắc xin mang tầm quốc tế tốn kém lắm, trong khi cơ chế chưa rõ ràng, Bộ Y tế còn có lúng túng; chỉ cấp phép khi thiếu vắc xin, còn đủ vắc xin thì không cấp phép, điều đó không hợp lý" - đại diện Nanogen nói.
Đại diện công ty này khẳng định kiến nghị trên "không nóng vội", xuất phát từ cơ sở khoa học là đánh giá từ Học viện Quân y và Viện Pasteur TP.HCM, hai đơn vị độc lập được Bộ Y tế chỉ định trực tiếp thử nghiệm. Đặc biệt, các kết quả thử nghiệm lâm sàng, khả năng sinh miễn dịch của vắc xin Nano Covax đạt 99,4%. Nếu so sánh với các loại vắc xin khác trên thế giới là không hề thua kém, thậm chí có phần cao hơn, trong khi giá bán "rẻ nhất thế giới" (120.000 đồng/liều).
Nanogen còn khẳng định trong quá trình thử nghiệm đều có báo cáo hằng ngày về Cục Khoa học công nghệ và đào tạo (Bộ Y tế). Các yếu tố về an toàn, sinh miễn dịch, hiệu quả bảo vệ đều thực hiện đáp ứng đúng hướng dẫn của Tổ chức Y tế thế giới.
"Vắc xin Nano Covax chỉ sản xuất phục vụ riêng cho người dân Việt Nam, không có nhu cầu bán ra nước ngoài. Khi có đủ miễn dịch cộng đồng, Nanogen sẽ ngừng để tập trung sản xuất các loại thuốc khác có giá trị thương mại cao hơn nhiều" - đại diện này nói và cho biết bên cạnh gửi kiến nghị cho Thủ tướng; đơn vị có đơn, thư gửi Bộ Y tế và bộ trưởng Bộ Y tế.
Tiêm Nano Covax trong ngày đầu tiên thử nghiệm lâm sàng giai đoạn 2 - Ảnh: NAM TRẦN
"Không thể 'nhảy cóc' trên tính mạng người dân"
Trao đổi với Tuổi Trẻ về vấn đề nêu trên, một thành viên trong hội đồng đạo đức của Bộ Y tế (trực tiếp tham gia phản biện quy trình thử nghiệm vắc xin Nano Covax) cho biết rất đồng thuận với ý kiến đánh giá của Bộ Y tế: nguyên tắc thử nghiệm lâm sàng một loại vắc xin bắt buộc phải hoàn tất 3 giai đoạn thử nghiệm.
"Không chỉ thiếu thời gian, vắc xin Nano Covax còn thiếu các số liệu khoa học cần thiết" - vị này khẳng định.
Vậy để đáp ứng việc cấp phép khẩn cấp, công ty cần thỏa điều kiện gì? Vị này cho biết trong các cuộc làm việc với công ty, hội đồng đạo đức của Bộ Y tế đã hướng dẫn rất cặn kẽ là phải hoàn thành thử nghiệm giai đoạn 3.
Cụ thể, hiện vắc xin này đã bước vào thử nghiệm giai đoạn 3 và phải hoàn thành thử nghiệm trên quy mô 13.000 người. Tất cả các mẫu lấy được phải chuyển cho một phòng thí nghiệm độc lập không có "xung đột" về lợi ích (ngoài Học viện Quân Y, Viện Pasteur) thực hiện. Sau khi có kết quả (an toàn, sinh miễn dịch, hiệu lực bảo vệ) mới thông qua hội đồng đạo đức của Bộ Y tế xem xét, nếu đủ điều kiện hội đồng sẽ tư vấn cấp phép.
"Ai cũng biết cần vắc xin gấp, Chính phủ và Bộ Y tế cũng rất khuyến khích sản xuất vắc xin trong nước nhưng về mặt khoa học, bất kỳ một loại vắc xin nào trên thế giới đều phải tuân thủ nguyên tắc đó. Không ai dám "nhảy cóc" trên sinh mạng người dân và với vai trò của hội đồng đạo đức chúng tôi phải phản biện, phải đặt tính mạng người dân lên trên hết" - vị này khẳng định.
Theo Bộ Y tế, đến nay Việt Nam đã phê duyệt có điều kiện cho 4 loại vắc xin COVID-19 của 4 hãng gồm Pfizer (Mỹ), AstraZeneca (Anh), Sputnik V (Nga), Sinopharm (Trung Quốc). Ngoài ra, vắc xin Moderna (Mỹ) cũng đang trong quá trình xem xét phê duyệt. Tất cả các loại vắc xin nói trên đều phải trải qua 3 giai đoạn thử nghiệm lâm sàng.
Cụ thể vắc xin của AstraZeneca thử nghiệm lâm sàng tại 11 quốc gia với 49.626 người tham gia; vắc xin của Sinopharm thử nghiệm lâm sàng tại 6 quốc gia với trên 45.000 người; vắc xin Sputnik V thử nghiệm lâm sàng tại 5 quốc gia với 21.977 người; vắc xin của Pfizer thử nghiệm lâm sàng tại 6 quốc gia với 43.418 người và vắc xin của Moderna thử nghiệm tại 4 quốc gia với 30.420 người.
Nghiên cứu sản xuất vắc xin chống COVID-19 tại Công ty cổ phần công nghệ sinh học dược Nanogen - Ảnh: DUYÊN PHAN
Hiểu thế nào về hai chữ "khẩn cấp"?
Với kiến nghị cấp phép khẩn cấp, đại diện Nanogen giải thích tức là xin cấp phép được sử dụng vắc xin tiêm chủng cho người dân, song song với việc thử nghiệm lâm sàng giai đoạn 3 cho đến khi đủ số lượng.
Trong khi đó, theo ông Trịnh Thanh Hùng - phó vụ trưởng Vụ Khoa học công nghệ các ngành kinh tế kỹ thuật (Bộ Khoa học và công nghệ), việc xem xét cấp phép cho vắc xin là thuộc thẩm quyền của Bộ Y tế nhưng trong cấp phép khẩn cấp, mỗi quốc gia có quy định riêng. Như ở Nga, Ấn Độ, Trung Quốc..., những thông tin ban đầu là căn cứ dữ liệu khi kết thúc thử nghiệm lâm sàng giai đoạn 2, cơ quan chức năng nước bạn đã cấp phép khẩn cấp và sau đó tiếp tục cho thử nghiệm giai đoạn 3 và theo dõi.
"Chúng tôi được biết Bộ Y tế đang xây dựng thông tư hướng dẫn việc cấp phép khẩn cấp. Việc được cấp phép khẩn cấp hay không phụ thuộc nhiều yếu tố, ý kiến một số nhà khoa học thì thấy được rồi. Qua tiêm thử nghiệm 1.000 người đầu tiên tham gia thử nghiệm lâm sàng giai đoạn 3 thấy cơ bản vắc xin được đánh giá là an toàn so với vắc xin khác" - ông Hùng nói.
Theo ông Hùng, ngay cả các vắc xin ngoại Việt Nam đã cấp phép cũng là cấp phép khẩn cấp và đã khẩn cấp thì vẫn phải theo dõi, đánh giá khi vắc xin được đưa vào sử dụng.
Về công nghệ, vắc xin Nano Covax sử dụng công nghệ protein tái tổ hợp, là công nghệ sản xuất được đánh giá an toàn. "Trong điều kiện thiếu vắc xin, thậm chí có tiền cũng không mua được, nhà sản xuất đặt vấn đề cấp phép sớm cho họ tôi thấy là đáng khích lệ. Tất nhiên việc có được hay không, khi nào cấp là thẩm quyền của Bộ Y tế" - ông Hùng nói thêm. L.ANH - H.LỘC
Quy trình cấp phép vắc xin của Việt Nam thế nào?
Theo Bộ Y tế, cũng giống như thông lệ quốc tế, tất cả vắc xin COVID-19 trước khi đưa vào sử dụng khẩn cấp tại Việt Nam đều phải tuân thủ thử nghiệm lâm sàng (thử nghiệm trên người tình nguyện) qua 3 giai đoạn, trên nguyên tắc đảm bảo 3 yếu tố: an toàn, sinh miễn dịch và quan trọng nhất là hiệu quả bảo vệ.
Trong đó, giai đoạn 3 được thực hiện trên quy mô lớn hơn. Kết quả của giai đoạn 3 sẽ quyết định việc vắc xin đó có được phê duyệt để triển khai tiêm chủng rộng rãi hay không. Sau khi hoàn tất 3 giai đoạn thử nghiệm lâm sàng, có báo cáo đánh giá, hội đồng đạo đức trong nghiên cứu y sinh của Bộ Y tế sẽ họp đánh giá kết quả nghiên cứu trước khi chuyển sang hội đồng tư vấn cấp giấy phép lưu hành thuốc, vắc xin và sinh phẩm y tế xem xét. Trong điều kiện hội đồng tư vấn cấp phép lưu hành thuốc, vắc xin và sinh phẩm y tế đồng thuận, sẽ báo cáo để bộ trưởng Bộ Y tế phê duyệt có điều kiện.
Trả lời Tuổi Trẻ, Thứ trưởng Bộ Y tế Trần Văn Thuấn cho biết: "Theo Luật dược, thông thường vắc xin thử nghiệm xong giai đoạn 3 và thành công mới xem xét cấp phép, nhưng trong trường hợp khẩn cấp (cụ thể là trong đại dịch), Bộ Y tế sẽ báo cáo Chính phủ, trình Quốc hội xem xét khi nghiên cứu đang ở giữa kỳ của giai đoạn 3 và có kết quả tốt".
Thời gian qua, ban đầu Bộ Y tế cho biết sẽ cho nghiên cứu theo hình thức "gối đầu" để rút ngắn thời gian sản xuất, nhưng thực tế có những thời điểm tiến độ nghiên cứu bị chậm do chờ đợi về hồ sơ, thủ tục, đặc biệt thời điểm bước vào giai đoạn 3 thử nghiệm lâm sàng. LAN ANH
Các nước cấp phép sử dụng khẩn cấp vắc xin ra sao?
Một số loại vắc xin của các nước
Mỹ, EU và Tổ chức Y tế thế giới (WHO) đòi hỏi các hãng dược phải có dữ liệu thử nghiệm lâm sàng giai đoạn 3, trong khi Nga và Trung Quốc đã cấp phép sử dụng khẩn cấp vắc xin COVID-19 trước khi các hãng dược có dữ liệu của giai đoạn 3.
Đánh giá cuốn chiếu
Theo trang web của Cơ quan Dược phẩm châu Âu (EMA), EMA đã tiến hành đánh giá cuốn chiếu (rolling review) dữ liệu của các hãng dược phẩm. Đây là biện pháp nhằm hoàn tất thủ tục đánh giá một loại thuốc hay vắc xin triển vọng trong trường hợp khẩn cấp về sức khỏe cộng đồng trong thời gian ngắn nhất có thể. Theo quy trình, các công ty phát triển vắc xin có thể thực hiện song song một số nghiên cứu nhất định thay vì làm lần lượt và có thể sử dụng mô hình thử nghiệm. EMA kiểm tra liên tục dữ liệu về tính an toàn và hiệu quả của vắc xin ngay khi vắc xin đang trong giai đoạn phát triển. Đến khi quá trình phát triển vắc xin đầy đủ, thủ tục đánh giá chính thức sẽ diễn ra rất nhanh vì dữ liệu đã được phân tích trước rồi.
Tại Mỹ, theo quy định của Cơ quan Quản lý thực phẩm và dược phẩm (FDA), để được cấp phép sử dụng khẩn cấp, hãng dược phải nộp tất cả dữ liệu an toàn giai đoạn 1, 2 và 3. Trong đó, dữ liệu giai đoạn 3 phải có thời gian theo dõi trung bình người tham gia thử nghiệm ít nhất là 2 tháng.
Từ đó, FDA sẽ đánh giá dữ liệu từ thử nghiệm lâm sàng, tư vấn cho các hãng dược về các tiêu chí của thử nghiệm lâm sàng của họ. Đồng thời, nêu các kỳ vọng của FDA về tính an toàn và hiệu quả của vắc xin trước khi hãng dược quyết định nộp đơn xin cấp phép sử dụng khẩn cấp.
Sau khi nộp đơn, FDA triệu tập một cuộc họp công khai của Ủy ban Tư vấn về vắc xin và sản phẩm sinh học liên quan (VRBPAC) để thảo luận về dữ liệu từ các thử nghiệm lâm sàng. Nếu đạt các tiêu chí của FDA, vắc xin sẽ được cấp phép sử dụng khẩn cấp.
Trong khi đó, WHO sẽ đánh giá nghiêm ngặt dữ liệu lâm sàng giai đoạn 2 và 3 cũng như các dữ liệu bổ sung về tính an toàn, hiệu quả, chất lượng và kế hoạch quản lý rủi ro liên quan đến vắc xin cần cấp phép. Cho tới nay, WHO đã cấp phép sử dụng khẩn cấp cho vắc xin COVID-19 của Hãng Pfizer/BioNTech (31-12-2020), AstraZeneca/Oxford (15-2-2021), Johnson & Johnson (12-3), Moderna (30-4), Sinopharm (7-5) và Sinovac (1-6).
Nga, Trung Quốc sử dụng khẩn cấp sau giai đoạn 2
Trung Quốc đã cho phép sử dụng các vắc xin COVID-19 chưa được phê duyệt do một số công ty trong nước phát triển trong trường hợp khẩn cấp từ ngày 22-7-2020. Đài CCTV ngày 23-8-2020 dẫn lời ông Zheng Zhongwei - trưởng ban chuyên gia tư vấn COVID-19 của Chính phủ Trung Quốc - cho biết việc cấp phép sử dụng khẩn cấp phù hợp với Luật quản lý vắc xin Trung Quốc.
Sau đó, Trung Quốc đã cấp phép sử dụng khẩn cấp có điều kiện đối với vắc xin Sinopharm vào ngày 30-12-2020. Theo Tân Hoa xã, kết quả sơ bộ từ các thử nghiệm lâm sàng giai đoạn 3 vắc xin cho thấy hiệu quả bảo vệ đạt 79,34%. Vào thời điểm tháng 8-2020, Sinopharm và Sinovac đang tiến hành các thử nghiệm lâm sàng giai đoạn 3 trên vắc xin COVID-19. Một trong những vắc xin của Sinopharm ngày 13-8-2020 đã chứng minh có tỉ lệ phản ứng bất lợi thấp với bệnh nhân tham gia thử nghiệm và có kết quả khả quan về tính sinh miễn dịch ở giai đoạn 1 và 2.
Trong khi đó, Nga cũng đã cấp phép sử dụng khẩn cấp cho vắc xin Sputnik V vào tháng 8-2020 chỉ sau khi thử nghiệm giai đoạn 1 và 2. Bộ trưởng Y tế Mikhail Murashko thời điểm đó cho biết vắc xin cho thấy "hiệu quả và an toàn cao" và không có tác dụng phụ nghiêm trọng. Tuy nhiên, theo trang sciencemag.org, giấy chứng nhận này chỉ cho phép tiêm vắc xin Sputnik V cho "một số lượng nhỏ cư dân của các nhóm dễ bị tổn thương", trong đó có nhân viên y tế và người già. Vắc xin Sputnik V không thể được tiêm rộng rãi cho đến 1-1-2021, sau khi các thử nghiệm lâm sàng quy mô lớn hơn được hoàn thành. ANH THƯ




Tối đa: 1500 ký tự
Hiện chưa có bình luận nào, hãy là người đầu tiên bình luận